Methods for analysis of simulation data
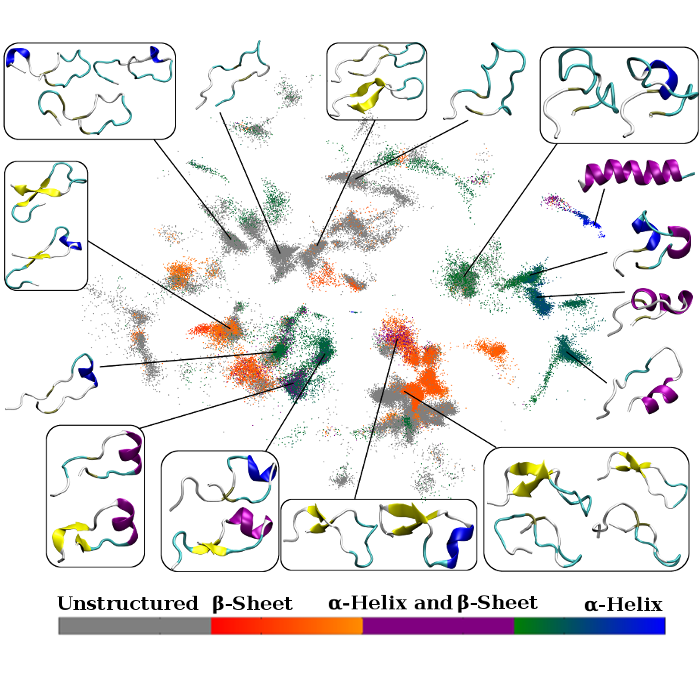
We focus on important computational issues that arise when assessing the results of MD simulations of various peptides and proteins and, more specifically, at the time of modeling their dynamical behavior, and when identifying their states and features and obtaining kinetic and thermo-dynamical estimates. We are applying and developing methods coming from machine learning. We use them for characterization of phase spaces of complex biological systems (dimensionality reduction techniques, clustering algorithms, etc.), as well as obtaining full kinetic and thermodynamical information (Markov State Models) in applications to intrinsically disordered proteins (i.e. α-synuclein, polyQ) and protein modifications (mono-ubiquitilated histone).