Bio-inspired materials

A controlled route towards biocompatible nanostructures and peptide-templated mineralization has immense relevance for drug delivery and tissue engineering. It is desirable to gain control over the understanding of parameters governing structural transformations and/or biomineralization process with an aim to generate nanostructures and artificial bone tissue of required morphologies and characteristics . Experimental methods are very useful to provide a macroscopic picture and are usually the first step towards understanding the intriguing details. The ability to fine-tune the morphology and control the characteristics of nanomaterials is highly desired. To do this an understanding of the response/influence of external factors (ionic strength, pH, etc.) at an atomistic-level is necessary. However, even with recent advancements, many times, experimental techniques alone are not sufficient to obtain such fine details. Classical MD simulations are relevant when the goal is to decipher details at an atomistic-level. However, classical MD, at times, is not capable due to time and length scale constrains. Therefore, we are utilizing multiscale simulation coarse-grained simulations and/or advanced sampling methods to understand the factors that control morphology in various systems, and biomineralization processes.
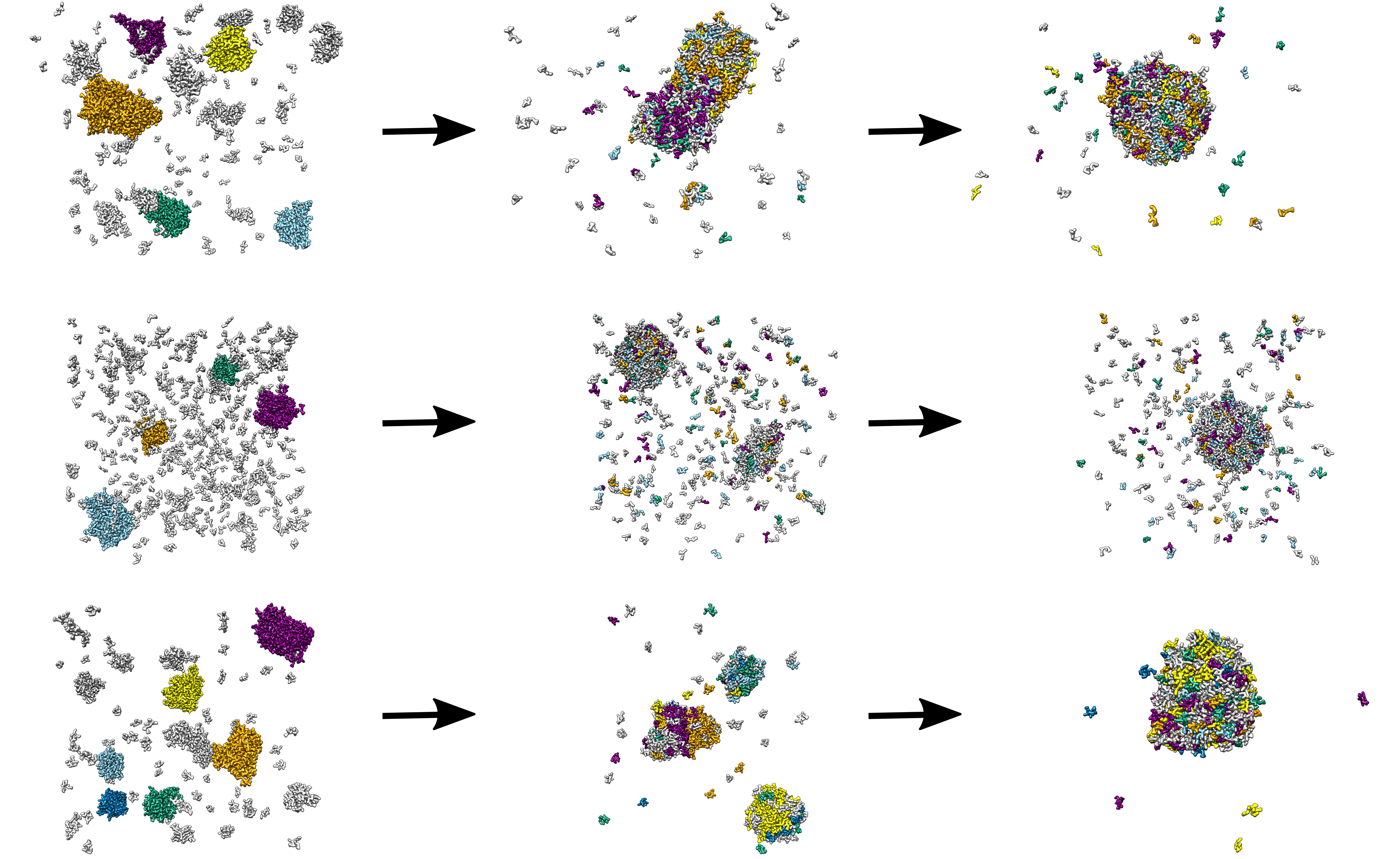
Development of new well ordered, functional biomaterials based on the underlying principal of self assembly has immense application in nanotechnology, nanomedicine and tissue engineering. Peptide based nano-materials are not only biocompatible but also their properties can be altered easily by slight changes in environmental conditions and/or side-chains of amino-acids. We are doing multiscale simulation study on many interesting peptides systems that exhibit different morphologies upon slightly altering the primary sequence of peptides.
J. Chem. Theory Comput. 2019, 15, 2, 1453–1462 doi

Biominerals are known to exhibit outstanding properties. Different morphologies and different shapes arranged in superstructures allow for a great variety of properties. How and when these structural features are encoded along the crystallization pathway is still largely unknown. While analyzing the macroscopic result of crystallization is quite easy, gaining insight into the early stages of crystallization through experimentation is difficult. The small size of these early stage structures, which is challenging for many experimental techniques on the one hand, is an advantage for molecular dynamics (MD) simulations on the other hand.