Multiscale simulation of viral capsids
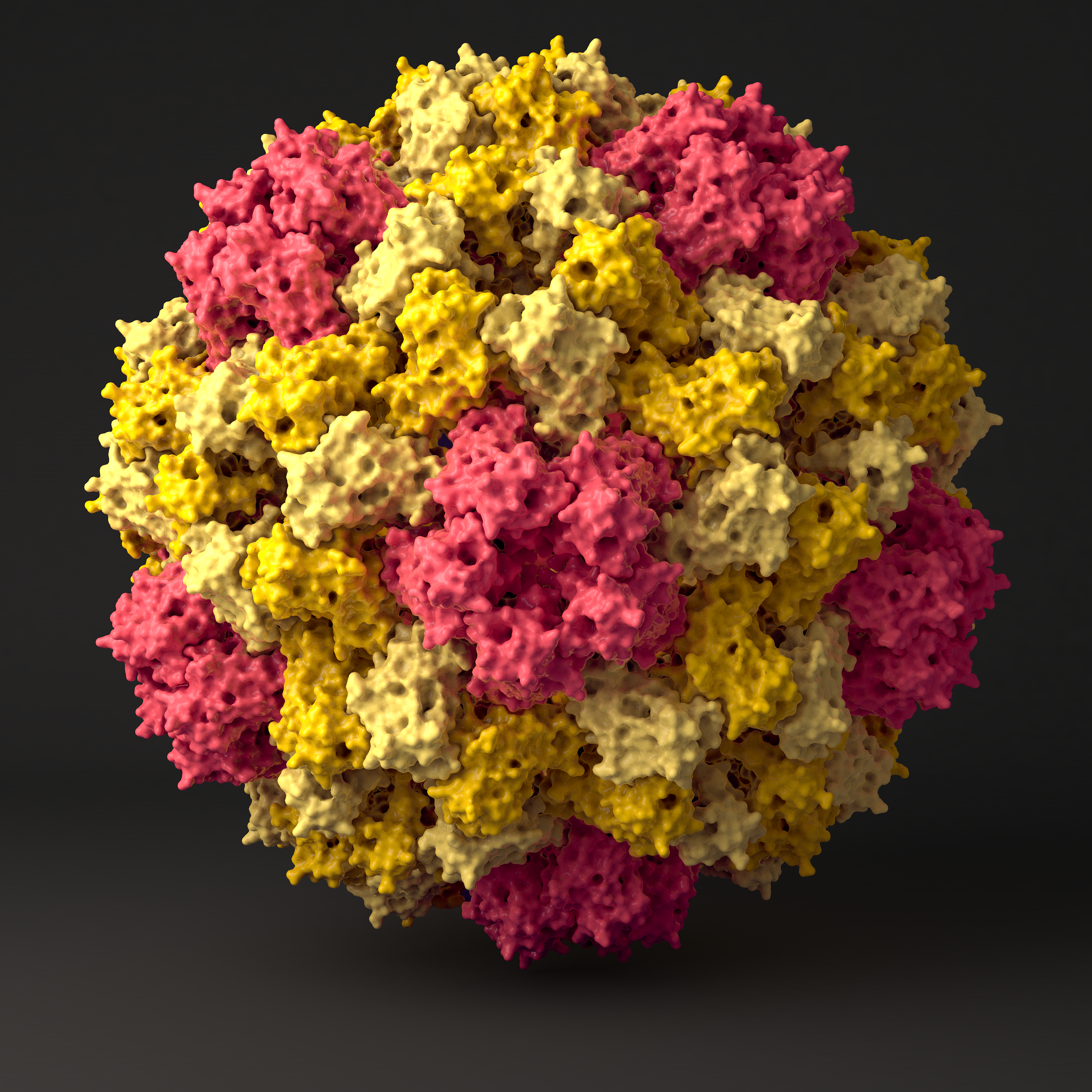
We use a combined approach of classical atomistic and different coarse grained (CG) simulation levels to investigate the 180-protein icosahedral capsid of Cowpea Chlorotic Mottle Virus (CCMV). First, the unstructured regions of the CCMV capsid proteins are studied by applying a suitable CG model together with clustering algorithms and free energy reweighting methods. The CG simulations combined with backmapping and subsequent atomistic simulations allow us to propose a multi-conformational ensemble for the experimentally- unresolved regions of the pentameric protein interface. In a second step, we use atomistic reference simulations to refine a CG protein model in such a way that it reproduces the elastic behavior of individual proteins and protein dimers in solution. The obtained model correctly predicts structural and elastic properties of bigger aggregates and mechanical properties of an entire virus capsid when compared to Atomic Force Microscopy experiment. Detailed analysis of the simulated rupture process allow us to propose an assembly model through well-defined oligomeric intermediate states, where the assembly order is regulated by the strengths of the interfacial binding, with a subsequent post-assembly reinforcement of weak spots by cooperative folding.