Biominerals

Biominerals are known to exhibit outstanding properties. Different morphologies and different shapes arranged in superstructures allow for a great variety of properties. It is desirable to gain control over the understanding of parameters governing structural transformations and/or biomineralization process. Experimental methods are very useful to provide a macroscopic picture and are usually the first step towards understanding the intriguing details. The ability to fine-tune the morphology and control the characteristics of nanomaterials is highly desired. Therefore an understanding of the response/influence of external factors (ionic strength, pH, etc.) at an atomistic-level is necessary. However, even with recent advancements, many times, experimental techniques alone are not sufficient to obtain such fine details.
Classical molecular dynamics simulations are relevant when the goal is to decipher details at an atomistic-level. However, classical MD, at times, is not capable due to time and length scale constrains. Therefore, we are utilizing multiscale simulation, coarse-grained simulations and/or advanced sampling methods to understand the factors that control morphology in various systems, and biomineralization processes.
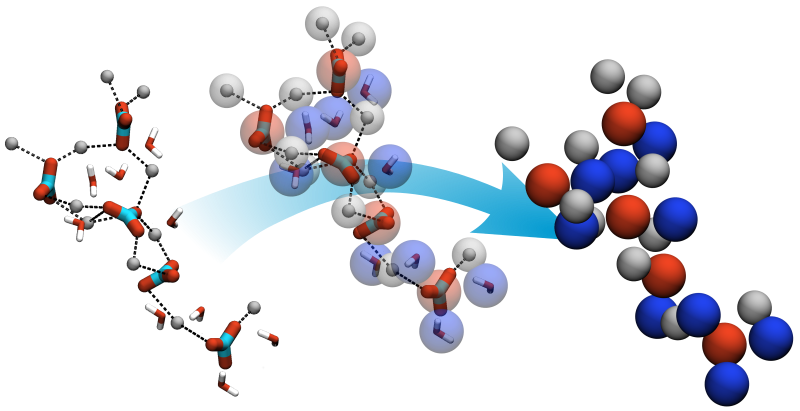
Development of a coarse-grained model for calcium minerals
Calcium minerals can be found in various materials, like teeth, bone and shells, but also be used or occur in industrial processes. The formation of these materials can be addressed by MD simulation on different resolution levels but is computational nevertheless expensive. For insights on the early stages of nanoparticle formation and growth we develop particle-based coarse-grained models for Calcium minerals.
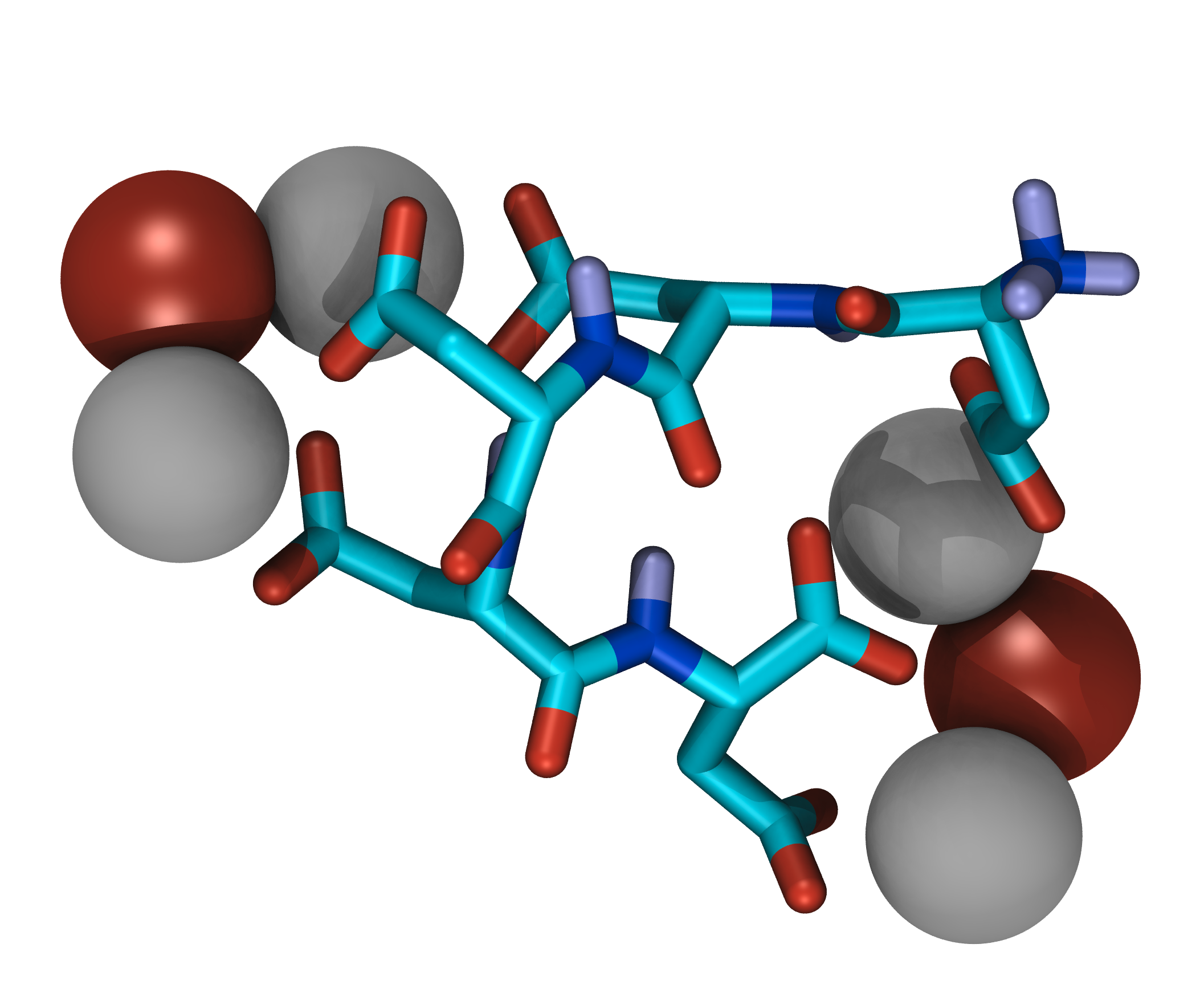
Mineralization modifiers
Some of the main components of biominerals besides the mineral itself are macromolecules, which appear to be the key for understanding their structural variety. Macromolecules isolated from calcium carbonate biominerals typically contain parts rich in aspartic acid. The influence of this amino acid on mineral formation can be explained with the ability to bind to metal ions with the negatively charged carboxylate group in the side chain.
Simulations of such mineralization modifiers suffer from slow sampling due to high energetic barriers caused by strong ion bridges. Even with the application of enhanced sampling techniques like Hamiltonian replica exchange and Metadynamics sampling to convergence remains a challenge. To tackle these sampling problems, the application of machine learning techniques is evaluated.
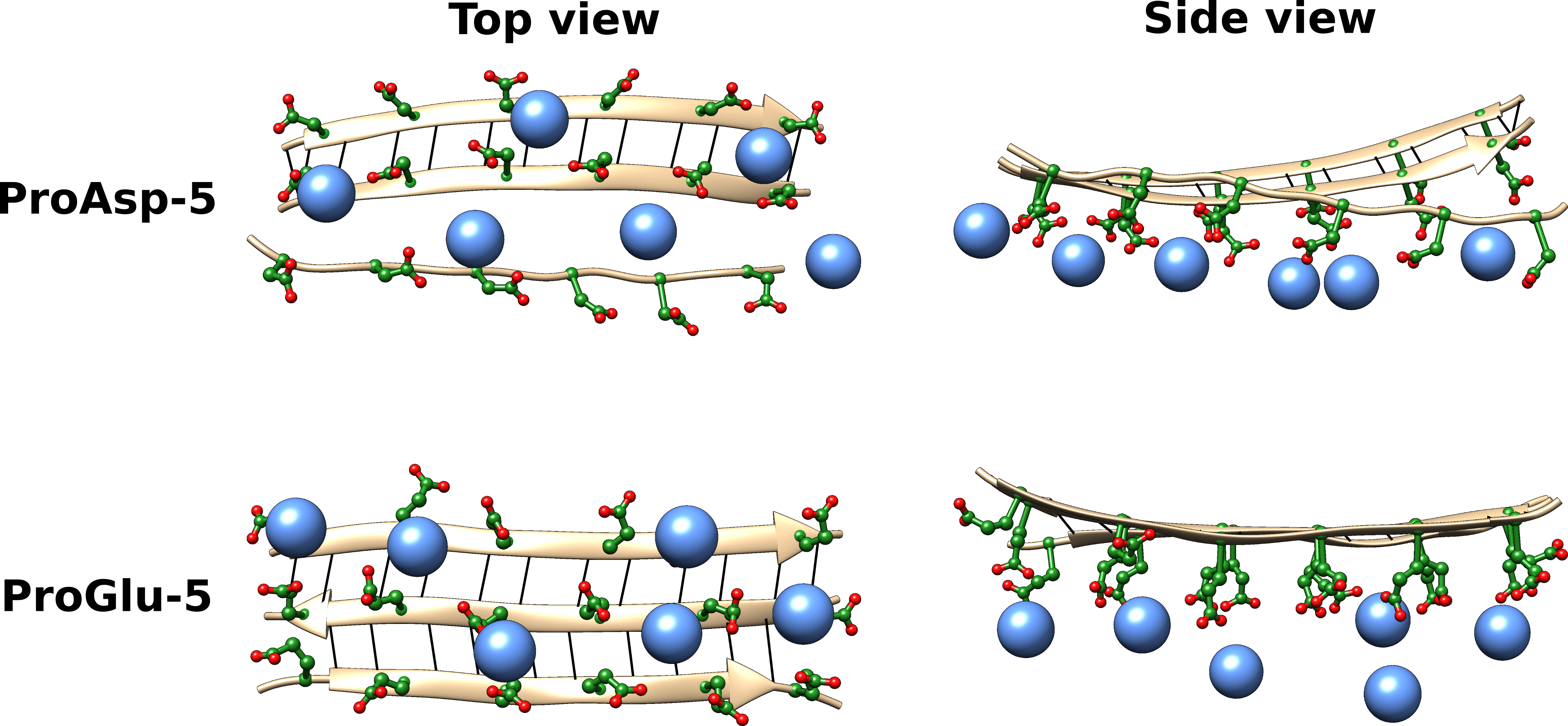
Biomineralization: Intrinsic details of the complex process
Biomineralization is the intricate process employed by living organisms to form minerals. Human orthopedic conditions, such as osteoporosis, are a direct consequence of poorly orchestrated biomineralization. Deciphering the molecular mechanism of this vital yet poorly understood process is thus essential for the development of therapeutic approaches. Using multiples simulations on amphiphilic peptides we identified the effects of various aspects of the peptide sequence (length, terminal, side-chains) on aggregate stability and ion-peptide interactions.
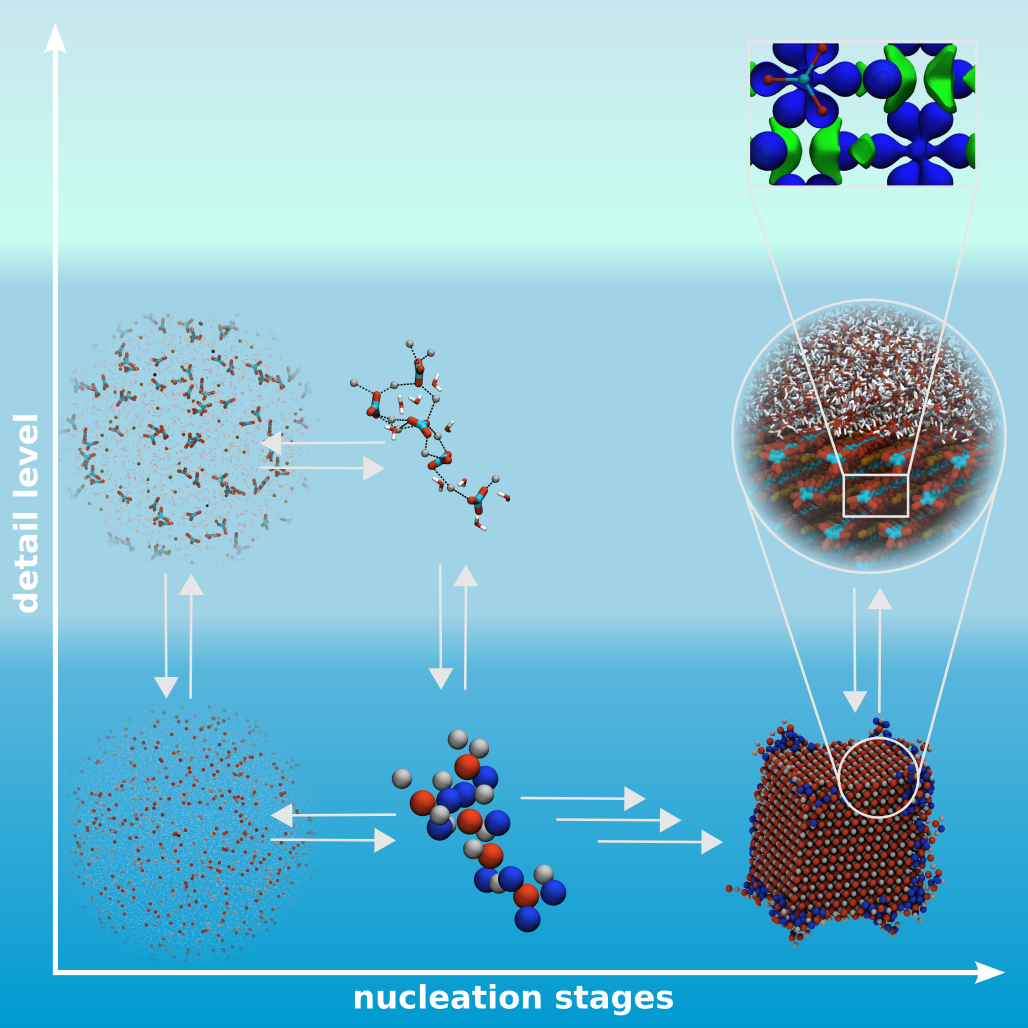
Multiscale nucleation models
Nucleation as well as crystallization and growth of nuclei are important steps in numerous processes and applications, found in the inanimate and animated world. It could be shown that these processes can follow different pathways, classical and non-classical nucleation, which can be addressed by MD simulations. However, the different stages of these processes are on complete different time and size scales. Therefore, studies on different resolution levels, coarse-grained, atomistic and quantum mechanic levels, are needed and have to be linked to give a insight view in the underlying mechanisms.
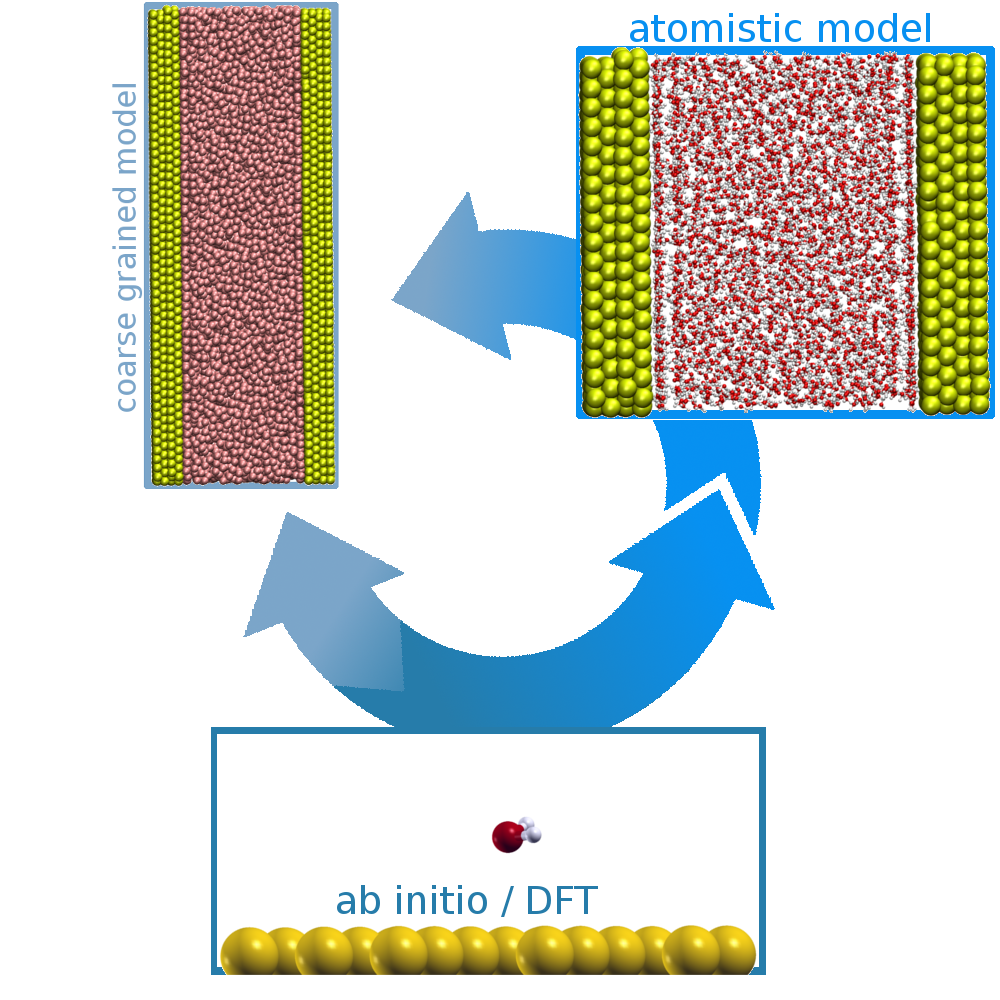
Solid-softmatter interfaces
Studying the behaviour of a liquid phase in the immediate vicinity of solid interfaces is a particularly active research field for material science. Molecular modelling is an outstanding tool for this kind of systems since it allows microscopic insight on the atomistic scale which is required to understand the complex interplay between heterogeneous systems.